
The U.S. Food and Drug Administration (FDA) doesn’t treat every generic drug application the same. Two different review paths exist - priority review and standard review - and choosing the right one can mean the difference between bringing a life-saving medication to market months earlier or waiting over a year. For generic drug makers, this isn’t just bureaucracy. It’s money, market share, and patient access.
What’s the difference between priority and standard review?
At its core, the difference is time. Standard review gives the FDA 10 months to decide if a generic drug application - called an ANDA (Abbreviated New Drug Application) - is approved. Priority review cuts that down to 8 months. That two-month window might sound small, but in the generic drug world, it’s huge.
Why does timing matter? Because the first company to get approval for a generic version of a brand-name drug gets 180 days of exclusive marketing rights. During that time, no other generic can enter the market. That exclusivity can generate hundreds of millions in revenue. A 2-month head start can mean capturing 90% of that first-year sales surge.
The FDA’s Office of Generic Drugs (OGD) handles all ANDAs. They don’t just flip a coin to decide which applications get priority. There are clear, published rules. If your drug meets one of these criteria, you qualify for priority review:
- You’re the first to submit a complete ANDA after the brand-name drug’s patents and exclusivity expire (a “first generic”).
- Your drug addresses a known shortage - like an antibiotic or insulin that’s hard to find.
- Your product is a complex generic that offers a real improvement over existing versions - say, a more stable formulation or better delivery system.
According to FDA’s 2022 data, 83.1% of priority review applications met their 8-month deadline. Standard review applications hit their 10-month target 72.3% of the time. That gap isn’t random - it reflects how much more complex many standard applications are.
How does the FDA decide who gets priority?
It starts before you even submit your application. The FDA’s Division of Filing Review checks your ANDA for completeness within 74 days. If anything’s missing - a missing bioequivalence study, unclear manufacturing details, incomplete labeling - you get a “Refuse-to-Receive” letter. That means you pay the $164,880 filing fee again and restart the whole process.
Once filed, your application enters the review clock. The FDA evaluates your drug across four areas: chemistry, manufacturing and controls (CMC), bioequivalence, and labeling. Most applications need more than one round of feedback. In 2022, 31.7% of ANDAs got a Complete Response Letter (CRL), meaning the FDA had concerns that needed fixing. Each round adds about 4.2 months to the timeline.
First generics are the easiest to qualify for priority review. The FDA’s Orange Book database tracks patents and exclusivity periods. If you’re the first to file after those protections expire, you’re almost guaranteed priority status - and the 180-day exclusivity that comes with it. In 2022, 92.7% of first generics received that exclusivity.
But here’s the catch: many drugs don’t have a clear “first generic” path. Some are complex - like inhalers, topical creams, or injectables with tricky formulations. These make up 18.3% of pending applications but only 9.7% of approvals. That’s why the FDA launched the Complex Generic Drug Product Pilot Program in January 2023. It lets companies get early feedback from FDA scientists before submitting. Companies using this program saw their first-cycle approval rate jump from 24.1% to 38.7%.
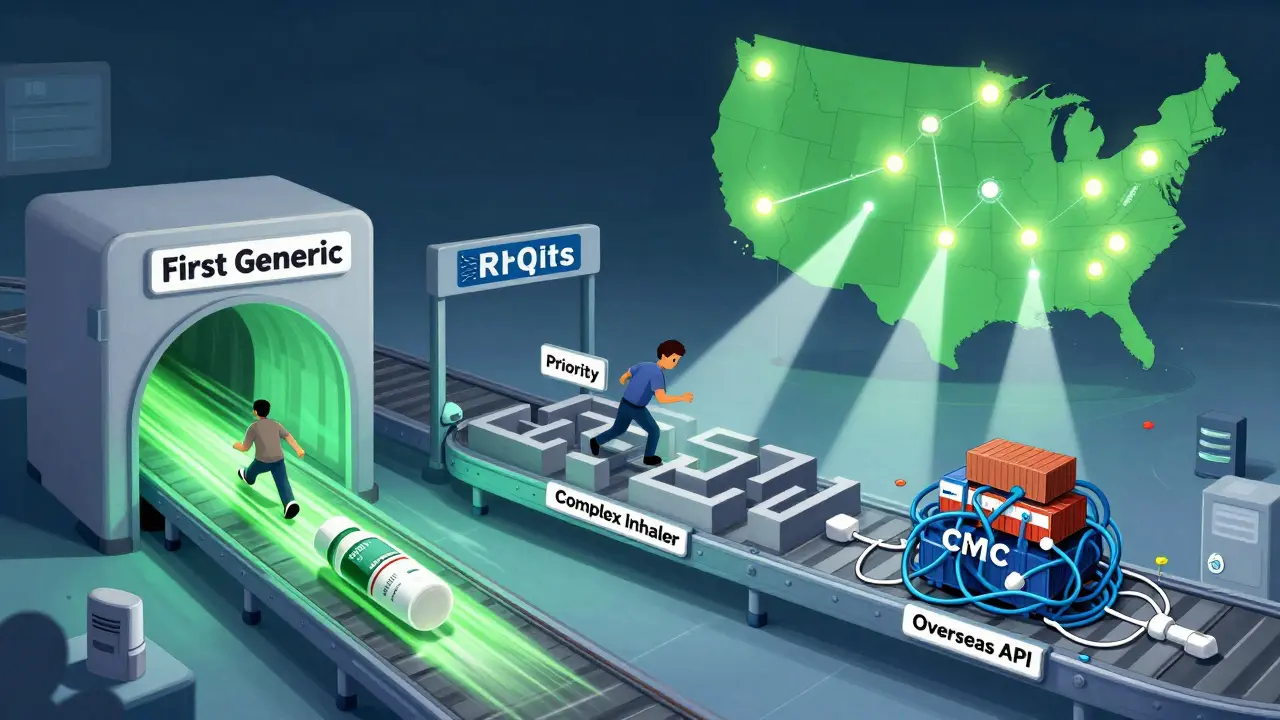
The new U.S. manufacturing push
In October 2023, the FDA dropped a major change: the ANDA Prioritization Pilot Program. This isn’t just about speed anymore - it’s about where drugs are made.
The new pilot gives priority review to ANDAs that meet all three of these criteria:
- Bioequivalence testing is done in the United States.
- The finished drug is manufactured in a U.S. facility.
- The active pharmaceutical ingredient (API) comes from a U.S.-based supplier.
This is a direct response to the pandemic. In 2021, the FDA reported that 80% of APIs were made overseas. When supply chains broke, shortages hit hard. Now, the FDA wants to fix that.
But it’s not easy. Only 12.3% of generic manufacturers currently meet all three requirements. Many rely on APIs from India or China. Complex excipients - the non-active ingredients that help the drug work - still mostly come from Europe. One regulatory manager on Reddit said, “The domestic manufacturing requirement is noble but unrealistic for complex generics.”
Still, companies are adapting. IQVIA data shows that 68% of major generic makers - like Teva, Sandoz, and Hikma - have increased their U.S.-based bioequivalence testing by 22% in the past year. Contract research firms like PPD and Covance are seeing a 35% jump in U.S. testing volume.
The goal? Raise the percentage of U.S.-manufactured generic drugs from 28% to 40% within five years. That’s ambitious, but it’s tied to the Biden administration’s broader push to secure pharmaceutical supply chains. The FDA estimates this pilot will help 45-60 applications per year get faster approval.
What’s changing behind the scenes?
The FDA isn’t just changing rules - it’s changing how it works. In 2023, the agency started testing AI tools to speed up review. In internal trials, AI helped cut review times for simple applications by 18.7%. By Q3 2024, those tools could be used on a wider scale.
More companies are also using pre-submission meetings. In 2020, only 41% of sponsors talked to the FDA before filing. Now, it’s 63%. That early dialogue helps avoid costly mistakes. One company told me they saved 8 months on a complex inhaler application by catching a labeling issue before submission.
And the numbers are climbing. In 2020, 21.1% of approved ANDAs got priority review. By 2022, that jumped to 28.4%. The FDA expects 32.8% of submissions in 2024 to be priority applications. Demand is growing - and so is competition.

Why does this matter to patients?
Generic drugs make up 88.6% of all prescriptions in the U.S. but only 15.3% of total drug spending. That’s how the system saves money - billions of dollars every year.
When a generic hits the market, brand-name prices drop fast - often by 80% or more. If a priority review gets a generic to market two months earlier, it means more patients get affordable access sooner. That’s especially critical for drugs used to treat diabetes, heart disease, or mental health conditions.
But delays still happen. On average, it takes 2.7 years from patent expiration to first generic approval. That’s too long. The FDA’s new tools and incentives are meant to shrink that gap. If they succeed, analysts predict an extra $18.7 billion in annual savings for the U.S. healthcare system by 2026.
What’s next for generic drug approvals?
The FDA’s next big move could be tighter integration of real-world data. Instead of relying only on lab-based bioequivalence studies, they might start using patient outcomes from electronic health records to confirm generic safety. That’s already being tested in Europe.
Also, expect more focus on quality over speed. The FDA’s 2023 report showed that 47.2% of CRLs were due to CMC issues - problems with how the drug is made. Better manufacturing standards, not faster approvals, will be the real game-changer.
For generic manufacturers, the message is clear: if you want priority review, you need to plan ahead. Start with the U.S. manufacturing requirements. Invest in pre-submission meetings. Build strong relationships with the FDA. And don’t assume your application will fly through the system - even priority reviews get CRLs if the data isn’t solid.
The system isn’t perfect. But it’s evolving - and it’s working. More generics are getting approved faster. More are being made here. And more patients are getting the medicines they need at prices they can afford.
What’s the main difference between priority and standard review for generic drugs?
The main difference is time. Standard review takes up to 10 months from submission to decision, while priority review is capped at 8 months. Priority review is reserved for first generics, drugs in shortage, or complex generics with clinical advantages. The faster timeline helps get affordable medicines to patients sooner.
Can any generic drug company apply for priority review?
No. Priority review is only available if the application meets specific criteria: being the first generic after patent expiration, addressing a drug shortage, or being a complex product with a meaningful improvement over existing versions. The FDA doesn’t grant priority review based on company size or request alone.
What is the ANDA Prioritization Pilot Program?
Launched in October 2023, this program gives priority review to ANDAs that use U.S.-manufactured active ingredients, U.S.-based finished drug production, and U.S.-conducted bioequivalence testing. It’s part of a broader effort to reduce reliance on overseas manufacturing and strengthen domestic supply chains.
How often do generic drug applications get rejected or delayed?
About 31.7% of original ANDAs received at least one Complete Response Letter (CRL) in 2022. Most delays come from chemistry, manufacturing, and controls (CMC) issues - like unclear processes or inconsistent quality data. Each CRL adds about 4.2 months to the timeline. Pre-submission meetings with the FDA can reduce these delays significantly.
How long does it usually take from patent expiration to a generic drug launch?
On average, it takes 2.7 years from the time a brand-name drug’s patent expires until the first generic is approved. This delay is often due to patent litigation, complex manufacturing requirements, or regulatory hurdles - even for priority applications. The FDA’s new tools aim to shorten this window.
Are U.S.-made generic drugs safer than those made overseas?
The FDA holds all approved generics - whether made in the U.S. or abroad - to the same strict quality and safety standards. The difference isn’t safety, but supply chain reliability. U.S. manufacturing reduces the risk of shortages caused by international disruptions, like pandemics or geopolitical issues.





8 Comments
So let me get this straight - we’re paying $164k just to get a letter saying ‘your paperwork’s messy’? And then we do it all over again? The FDA’s filing system is like a vending machine that eats your money and spits out a napkin.
/p>But hey, at least the 8-month clock is faster than my Wi-Fi on a Monday morning. Two months might not sound like much, but in generic drug time? That’s a whole season of Netflix. And if you’re the first one in? Congrats, you just won the lottery with a side of insulin.
This is the most important thing no one’s talking about. We’re letting India and China control our medicine supply while we sit around waiting for our ‘priority’ review like it’s a VIP pass to a concert that’s sold out. If you’re not making the API in the U.S., you’re not part of the solution - you’re part of the problem.
/p>Stop pretending ‘quality standards’ are the same across borders. The FDA can inspect factories in the U.S. every week. Can they do that in Mumbai? In Shanghai? Not even close. This pilot isn’t ‘unrealistic’ - it’s overdue. If we want to stop dying because a shipment got stuck in a port, we need to make drugs here. Period.
Let’s be honest - the entire system is a performance. The FDA claims they’re ‘streamlining’ reviews, but 31.7% of applications get a CRL? That’s not inefficiency - that’s intentional gatekeeping. They want you to burn cash on consultants, delay for months, and then ‘finally’ get approved after paying for three rounds of revisions.
/p>And don’t get me started on the ‘first generic’ loophole. Big pharma sits on patents for 15 years, then suddenly someone files on day one? That’s not innovation - that’s legal arbitrage. The FDA rewards lawyers, not scientists. And now they’re rewarding U.S. manufacturing? Cute. But only if you’re already a billion-dollar company with a D.C. lobbyist. The little guys? They’re still stuck in the same mud.
The structural challenges in global pharmaceutical supply chains are multifaceted and cannot be resolved through unilateral policy interventions alone. While domestic manufacturing initiatives are commendable from a national security perspective, they risk creating inefficiencies in cost structures that disproportionately impact low-income populations who rely on affordable generics.
/p>Moreover, the current regulatory framework, though rigorous, lacks harmonization with international standards such as those established by the WHO and ICH. A more sustainable approach would involve capacity-building in emerging manufacturing hubs, rather than imposing geographic restrictions that may inadvertently reduce overall market competition and access.
It is also worth noting that bioequivalence testing conducted abroad, when performed under accredited laboratories, yields data of comparable scientific validity to that generated domestically. The emphasis should thus be on data integrity, not location.
Priority review? More like priority to the companies that can afford to hire 12 lawyers and a team of regulatory consultants just to fill out the form right.
/p>And let’s not pretend the FDA’s AI tools are going to fix anything. They’re just automating the same bureaucratic nonsense with more buzzwords. If you think a machine can understand why a 37-page CMC section is ‘incomplete,’ you’ve been watching too many Silicon Valley documentaries.
Also, ‘first generic’ exclusivity? That’s just a 180-day monopoly dressed up as public policy. The real winners aren’t patients - they’re the shareholders of the company that filed first. And the FDA? They’re just the bouncer letting the VIPs in.
U.S.-made API? Cool. Now tell me how I’m supposed to afford my blood pressure med when it costs $150 instead of $4. 🤡
/p>Also, why is the FDA suddenly obsessed with geography? Is the medicine less effective if it’s made in Bangalore? Or is this just a fancy way of saying ‘buy American’? Because if so, congrats - you just turned healthcare into a political slogan.
And AI? Yeah, I’m sure the algorithm will understand why my tablet’s dissolution profile is ‘non-comparable’ after 3 a.m. on a Friday. 😴
There’s a quiet revolution happening in the background of these regulatory changes. More companies are engaging in pre-submission meetings - and it’s making a real difference. One manufacturer I spoke with reduced their review time by 8 months simply by clarifying labeling requirements before filing.
/p>It’s not about rushing approvals. It’s about reducing friction. The FDA’s shift toward early dialogue, AI-assisted triage, and transparency in feedback is slowly turning a slow-moving bureaucracy into a more responsive system.
Yes, the U.S. manufacturing goals are ambitious. But if even 40% of generics are made domestically in five years, it’s a win for resilience - not just patriotism. The goal isn’t to exclude global partners; it’s to build redundancy. And that’s smart policy.
Broooooo. The FDA is acting like they’re the boss of the whole world. You think I’m gonna cry because my insulin isn’t made in Ohio? My cousin in Lagos is still waiting for his meds because the FDA said ‘API not approved’ - but the same drug was made in India and sold in Kenya for 3 years.
/p>Y’all are turning healthcare into a Netflix drama. ‘The Rise of the American Pill.’
Meanwhile, real people? Still waiting. 😭